

The patient was admitted to the hospital for continued neurological workup. Blood samples were sent to an outside lab to check for the presence of anti-acetylcholine (AcH) and anti-muscle-specific tyrosine kinase (MuSK) antibodies. Soon after arriving to the floor, the patient was evaluated by the staff neurologist, who agreed with our diagnosis and ordered prednisone and pyridostigmine. A chest CT demonstrated no thymic or respiratory abnormalities, and brain MRI was unremarkable. Approximately 24 hours after first arriving in the ED, the patient reported that his symptoms had greatly improved and exhibited no ptosis or vertical gaze deficit.
Discussion
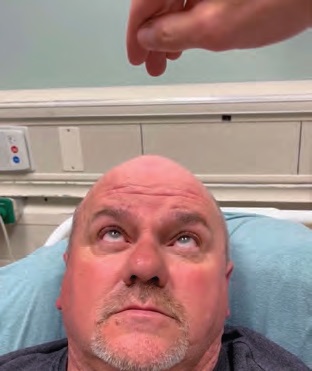
IMAGE 5: Upward gaze after ice pack test.
Myasthenia gravis is the most common disorder of neuromuscular transmission.2 The prevalence of MG in the United States is estimated to be 37 per 100,000, with estimated incidence ranging from 4.1 to 30 cases per million person-years.3,4 The disorder most commonly initially presents in females more than 40 years old and men over the age of 60. MG may present at any age in either sex, although it is rare in children.3 MG has a strong association with thymic hyperplasia and thymoma. It is estimated that 13 percent of patients with MG have one or more coexisting autoimmune diseases, most commonly affecting the thyroid gland. The estimated rate of co-occurrence is 7 percent with Graves’ disease and 3 percent with Hashimoto’s thyroiditis, which may contribute to symptoms of ophthalmopathy and weakness if present.5
Most patients initially present with intermittent flares of ptosis, extraocular muscle weakness, and diplopia. The second most common presentation involves bulbar muscle weakness, dysphagia, dysarthria, and frequent aspiration.6 A key feature in diagnosis is weakness that worsens with muscle use and improves with rest. Left untreated, weakness may spread and become generalized. In severe flares, known as myasthenic crises, the diaphragm and accessory muscles of breathing may become significantly weak, requiring noninvasive positive airway ventilation or intubation with mechanical ventilation. For this reason, prompt diagnosis and initiation of treatment are vital for individuals experiencing their first MG flare.
The mechanism in ptosis and in MG is believed to be the inhibition of the levator palpebrae superioris muscle at its lower motor neuron synapse with the oculomotor nerve. This is caused by the blockade of nicotinic AcH receptors by autoantibodies. Several mechanisms have been proposed to explain the temporary improvement of ocular MG symptoms after cooling.1 Although colder temperatures have been demonstrated to decrease the speed of nerve conduction, they also inhibit the action of acetylcholinesterase, resulting in a greater amount of neurotransmitter for a longer period to be present in the gap junction. This mechanism is also applied in the use of the acetylcholinesterase inhibitor pyridostigmine as first-line treatment for myasthenic flares.6,7
Pages: 1 2 3 4 | Single Page



No Responses to “Case Report: The Ice Pack Test”